6. 核心过滤模块
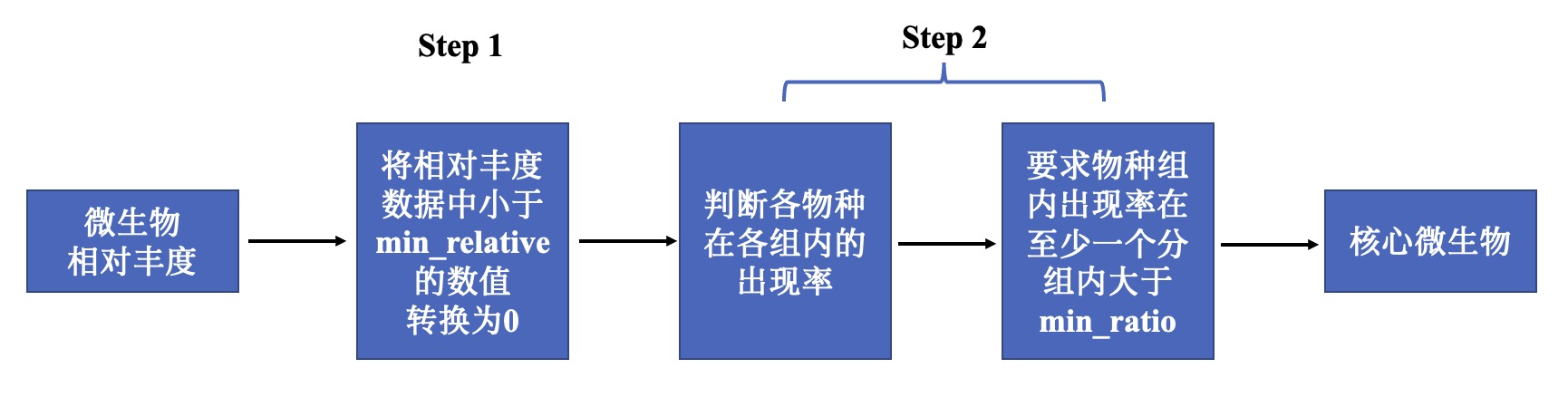
在微生物注释分析结果中,可以发现存在相当多的”稀有物种“。这些”稀有物种“具备相对丰度或者在样本内出现率较低的特点,对于筛选组间微生物差异性造成了极强的干扰。特别是筛选关键微生物的分析中,机器学习算法,例如随机森林、LEFse,很容易将这些个别组内存在的稀有物种,识别为组间的差异物种,因此有必要在正式分析前将这些稀有物种根据统一的标准进行过滤。本章节的data_filter模块,根据物种相对丰度和物种组内出现率对物种进行过滤,并筛选出不同注释级别的核心微生物。过滤的基本流程为:首先将在不同注释级别中将全部出现的物种进行编号,再根据设定的最小物种相对丰度使低于此阈值的丰度转换为0,最后将要求核心物种必须满足在至少一个分组内物种组内出现率高于预先设定的阈值,其余物种则判断为”稀有物种“进行过滤。
6.1 data_filter模块
data_filter模块可以将自动识别输入微生物数据的物种注释级别,并根据物种相对丰度和物种组内出现率筛选出核心微生物物,支持dataframe、list和file三种读取方式。
6.1.1参数介绍
data输入微生物数据支持
dataframe和list两种格式,dataframe应符合3.1中的格式要求,也可将不同级别微生物数据dataframe直接合并为list输入。dir:指定微生物数据文件存放的地址,文件可以以
csv和txt格式保存,格式应符合3.1中的格式要求。min_relative:指定最小的微生物物种相对丰度数值,低于此阈值的相对丰度将会被过滤为0。
min_ratio:指定最小的微生物物种在至少一个分组内的出现率,低于此阈值的物种将会被过滤。
design:指定样本分组信息
mapping文件,支持直接输入dataframe或者指定文件地址。adjust:在
min_relative和min_ratio过滤下,部分样品的微生物物种可能过滤为空,打开此选项可以将空样本赋予一个特殊特征在beta多样性中以区分彼此。【默认:False】pattern:指定在文件夹内微生物数据文件名的特征信息,与
dir配合使用output:指定是否将核心微生物计算结果输出。【默认:False】
change:Qiime2产生物种注释结果中有时出现完全为空的情况(
d__Bacteria;__;__),打开此选项后可以将其修正为(d__Bacteria;Other;Other)。【默认:False】change_name:指定空注释的修改名,与
change配合使用。【默认:Other】
6.1.2 使用范例
数据基本读取方式
代码示例一 : 通过文件Files读取
将符合3.1和3.1.3要求的微生物数据及分组信息文件放置在工作目录中
Tips1: 可以根据实验分析情况设置不同分组或者样本数量的mapping文件,EMP包将自动根据mapping文件从微生物数据中提取样本,无需手动调整。
Tips2:可以利用 pattern参数选择自己想要的微生物数据。例如当示例数据是按照L2-L7对微生物文件进行命名,如只需要分析种级别数据,则可以指定pattern='L7',这样函数将只会读取种级别数据进行分析。
Tips2:如果不需要对微生物数据进行过滤,可以设置min_relative = 0和min_ratio = 0
library(EasyMicroPlot) # 加载包
core_data <- data_filter(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7,pattern='txt')
# 核心物种(门级别)
core_phylum <- core_data$filter_data$phylum
core_phylum_ID <- core_data$filter_data$phylum_ID
# 核心物种(种级别)
core_species <- core_data$filter_data$species
core_species_ID <- core_data$filter_data$species_ID
代码示例二 :通过数据框读取
library(EasyMicroPlot)
raw_species <- read.table('16s_data/otu_table_even_L7.txt',header = T)
mapping <- read.table('mapping/mapping.txt',header = T)
core_data <- data_filter(data=raw_species,design = mapping,
min_relative = 0.001,min_ratio = 0.7 )
# 核心物种(种级别)
core_species <- core_data$filter_data$species
core_species_ID <- core_data$filter_data$species_ID
代码示例三 :通过列表读取
library(EasyMicroPlot)
file1 <- read.table('16s_data/otu_table_even_L2.txt',header = T)
file2 <- read.table('16s_data/otu_table_even_L7.txt',header = T)
mapping <- read.table('mapping/mapping.txt',header = T)
file_list <- list(file1,file2)
core_data <- data_filter(data=file_list,design = mapping,min_relative = 0.001,min_ratio = 0.7 )
# 核心物种(门级别)
core_phylum <- core_data$filter_data$phylum
core_phylum_ID <- core_data$filter_data$phylum_ID
# 核心物种(种级别)
core_species <- core_data$filter_data$species
core_species_ID <- core_data$filter_data$species_ID
数据结果展示
# core_data$filter_data 过滤后样本各个级别的核心微生物结果
core_phylum <- core_data$filter_data$phylum
core_phylum # 门水平核心微生物数据
core_phylum_ID <- core_data$filter_data$phylum_ID
core_phylum_ID #门水平核心微生物详细注释信息