12. 随机森林交叉验证模型
近年来主流的微生物分析中,随机森林模型常被用来进行关键微生物的筛选。但是由于随机数导致模型结果的不确定性以及难以确定最优价值的差异菌种,因此随机森林交叉验证递归剔除弱重要性的策略可以帮助研究者在一定程度下筛选出一批具有潜在重要性的关键菌种。本章节RFCV模块提供了一种快速根据核心微生物进行各个级别随机森林模型快速筛选的方法及可视化结果。
12.1 RFCV模块
RFCV模块是可以根据4.1的data_filter函数筛选出各个级别的核心微生物并进行随机森林交叉验证模型筛选特征微生物物种。
12.1.1 参数介绍
RF由
data_filter函数产生的包含各个样本微生物的数据框。seed_start此参数将指定起始种子。【默认:123】
ntree每个随机森林的建树数目。
kfold交叉验证的数目。
rep建模重复数目。【默认:10】
RF_importance设定模型判断重要性的方式 (MeanDecreaseAccuracy, MeanDecreaseGini),对应值为1和2。【默认:1】
step设定剔除特征的步伐数目。【默认:1】
each_ouput设定是否输出每个随机种子下随机森林的结果图
x_break设定RFCV图形横轴的分割距离。
cutoff_colour设定RFCV图形截线的颜色。
palette指定绘图色板。
12.1.2 使用范例
代码示例:
# 首先利用过滤核心模块读取原始数据
library(EasyMicroPlot) # 加载包
core_data <- data_filter(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7)
# 选择出自己需要的微生物级别数据
RF_data <- core_data$filter_data$species
## 进行随机森林交叉验证模型计算
RF_re <- RFCV(RF_data)
RF_re$RFCV_data # 输入的微生物数据
RF_re$RFCV_result #随机森林交叉验证的基本结果
RF_re$RFCV_result_plot #机森林交叉验证的图形结果
基本计算结果:
# 输入的微生物数据,这里默认采用最常用的种级别数据作为结果展示,用户可以自行选择需要的级别
RF_re$RFCV_data
# 读取模型计算过程中的准确率变化趋势
RF_var_dataframe <- data.frame(matrix(unlist(RF_re$RFCV_result$CV_accuracy),nrow = c(ncol(RF_data)-2)))
seed_name=RF_re$RFCV_result$Seed_num # 获得模型计算时的种子
colnames(RF_var_dataframe)=paste( "Seed_", seed_name, sep="") # 修改列名
RF_var_dataframe
| Seed_123 | Seed_124 | Seed_125 | Seed_126 | … | Seed_129 | Seed_130 | Seed_131 |
|---|---|---|---|---|---|---|---|
| 0.8 | 0.85714286 | 0.80357143 | 0.91666667 | … | 0.8 | 1 | 0.71666667 |
| 0.8 | 0.85714286 | 0.875 | 0.91666667 | … | 0.8 | 1 | 0.8 |
| 0.8 | 0.85714286 | 0.80357143 | 0.91666667 | … | 0.8 | 1 | 0.71666667 |
| 0.8 | 0.85714286 | 0.80357143 | 0.91666667 | … | 0.8 | 1 | 0.8 |
| 0.8 | 0.85714286 | 0.80357143 | 0.79166667 | … | 0.8 | 1 | 0.9 |
| ... | ... | ... | ... | … | ... | ... | ... |
| ... | ... | ... | ... | … | ... | ... | ... |
| 0.55 | 0.92857143 | 0.875 | 0.875 | … | 0.9 | 1 | 0.81666667 |
| 0.71666667 | 0.85714286 | 0.80357143 | 0.91666667 | … | 0.775 | 0.83333333 | 0.81666667 |
| 0.7 | 0.82857143 | 0.67857143 | 0.91666667 | … | 0.675 | 0.66666667 | 0.91666667 |
图形结果展示:
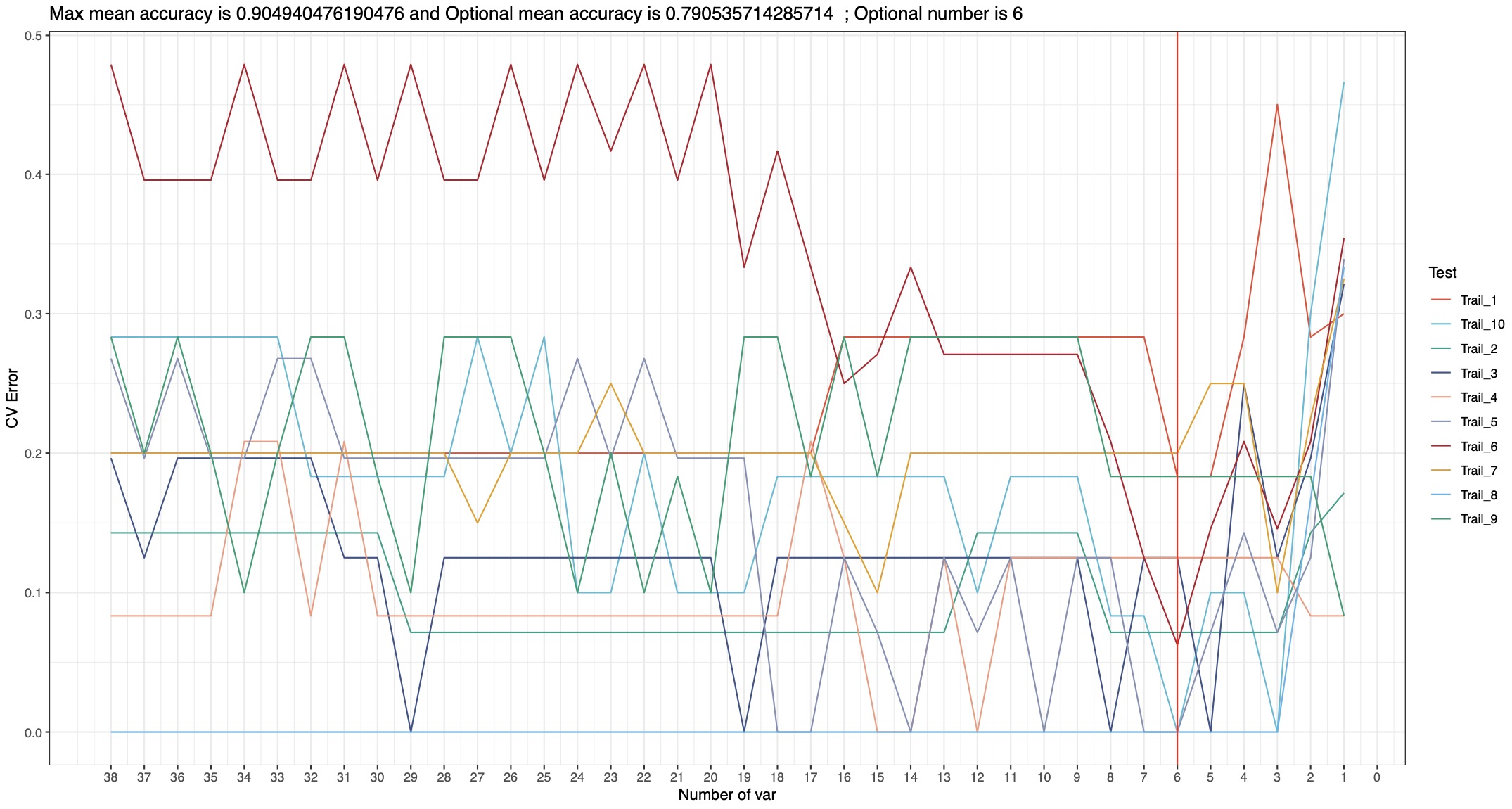
Tips 1: 下图所示截线依据10次随机数种子下平均错误率和错误率标准差最小的标准而选取,即意味着在这个截线下的特征数目可以保证10次随机森林交叉验证结果的平均错误率相对最低且在不同随机数种子下错误率相对稳定。
# RFCV平均错误率下降图
RF_re <- RFCV(RF_data)
RF_re$RFCV_result_plot$curve_plot
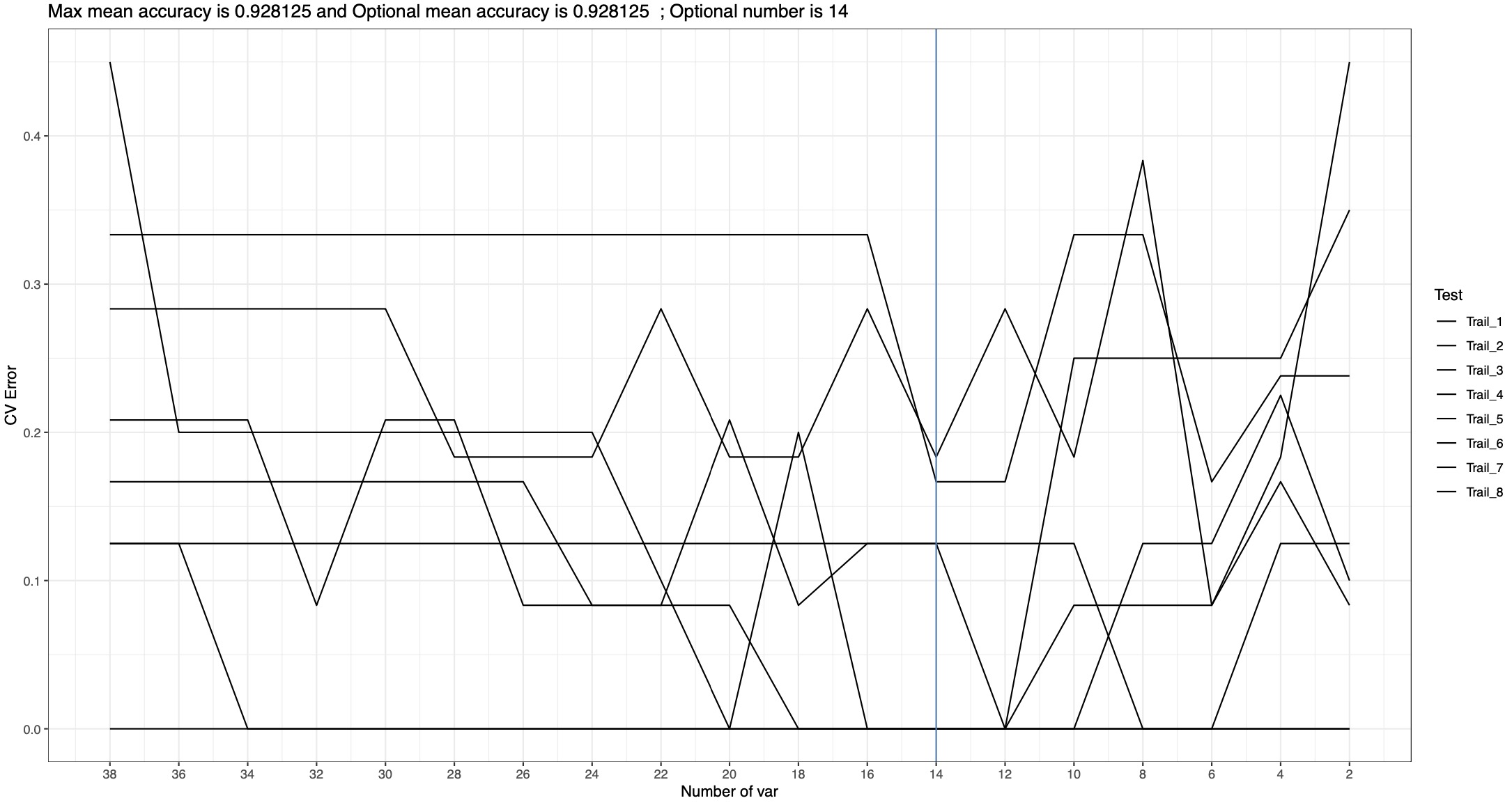
 Tips 2: 注意模型参数调整后,最适结果也将发生变化。
Tips 2: 注意模型参数调整后,最适结果也将发生变化。
# 也可以通过修改内置参数,调整模型
# 将步伐调整为2,横坐标调整为2,截线调整为蓝色,整体色板统一为黑色,模型重复数为8,起始种子为123
RF_re2 <- RFCV(RF_data,step = 2,x_break = 2,cutoff_colour = 'steelblue',
palette = 'black',rep = 8,seed=123)
RF_re2$RFCV_result_plot$curve_plot
# 由于不同随机数种子结果不同,因此需要判断该如何选择特征物种
# RFCV提供了不同随机数种子下最优特征数的交集和并集
# 交集
RF_re$RFCV_result_plot$intersect_num
[1] "V19" "V83" "V143" "V63"
# 并集
RF_re$RFCV_result_plot$union_num
[1] "V19" "V83" "V143" "V63" "V121" "V15" "V88" "V28" "V67"
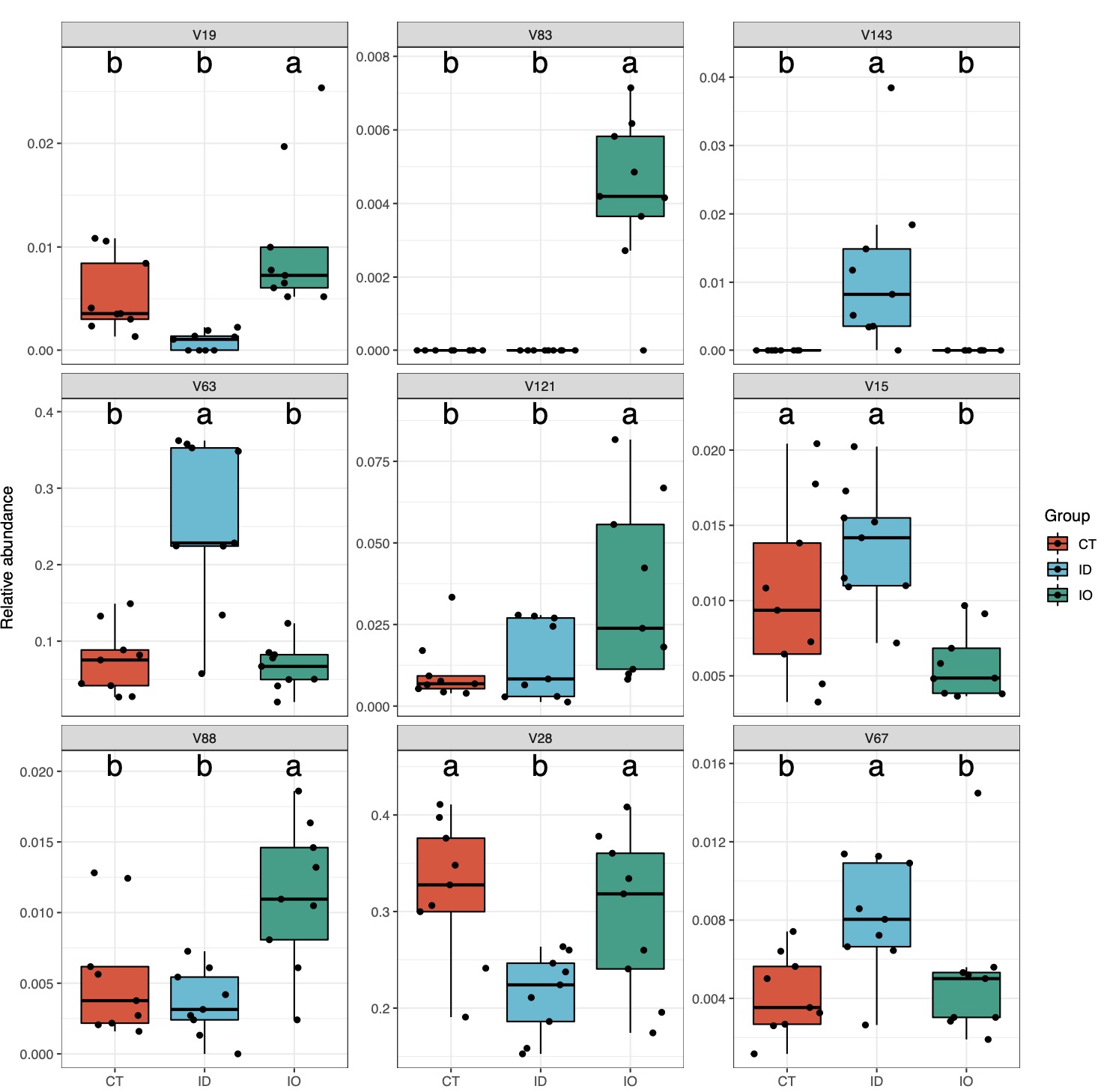
Tips 2: 由于随机森林判断的最优特征不一定具有统计学意义,因此可以利用tax_plot函数快速查看各组间差异结果
# 快速计算潜在关键菌的统计学差异
tax_re <- tax_plot(RF_data,tax_select = RF_re$RFCV_result_plot$union_num,method = 'LSD')
tax_re$pic$total
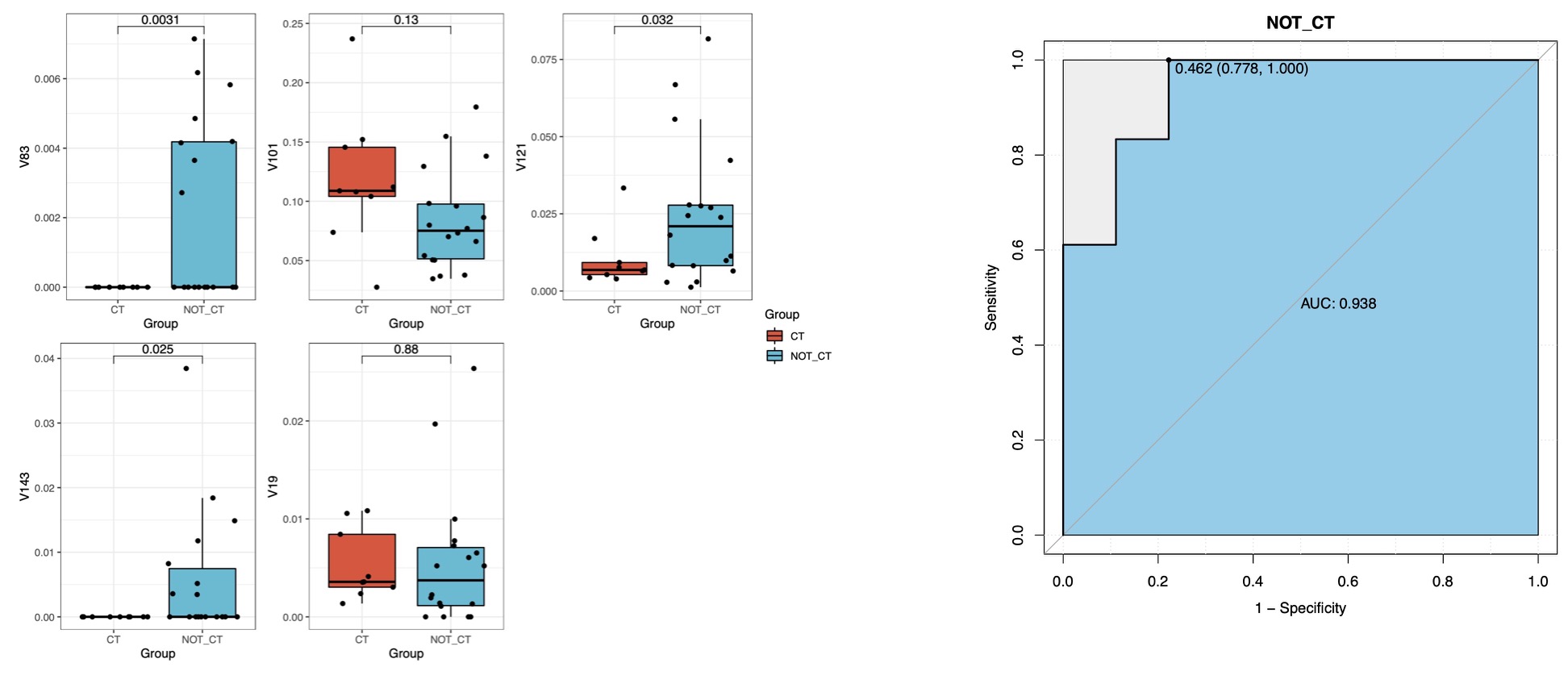
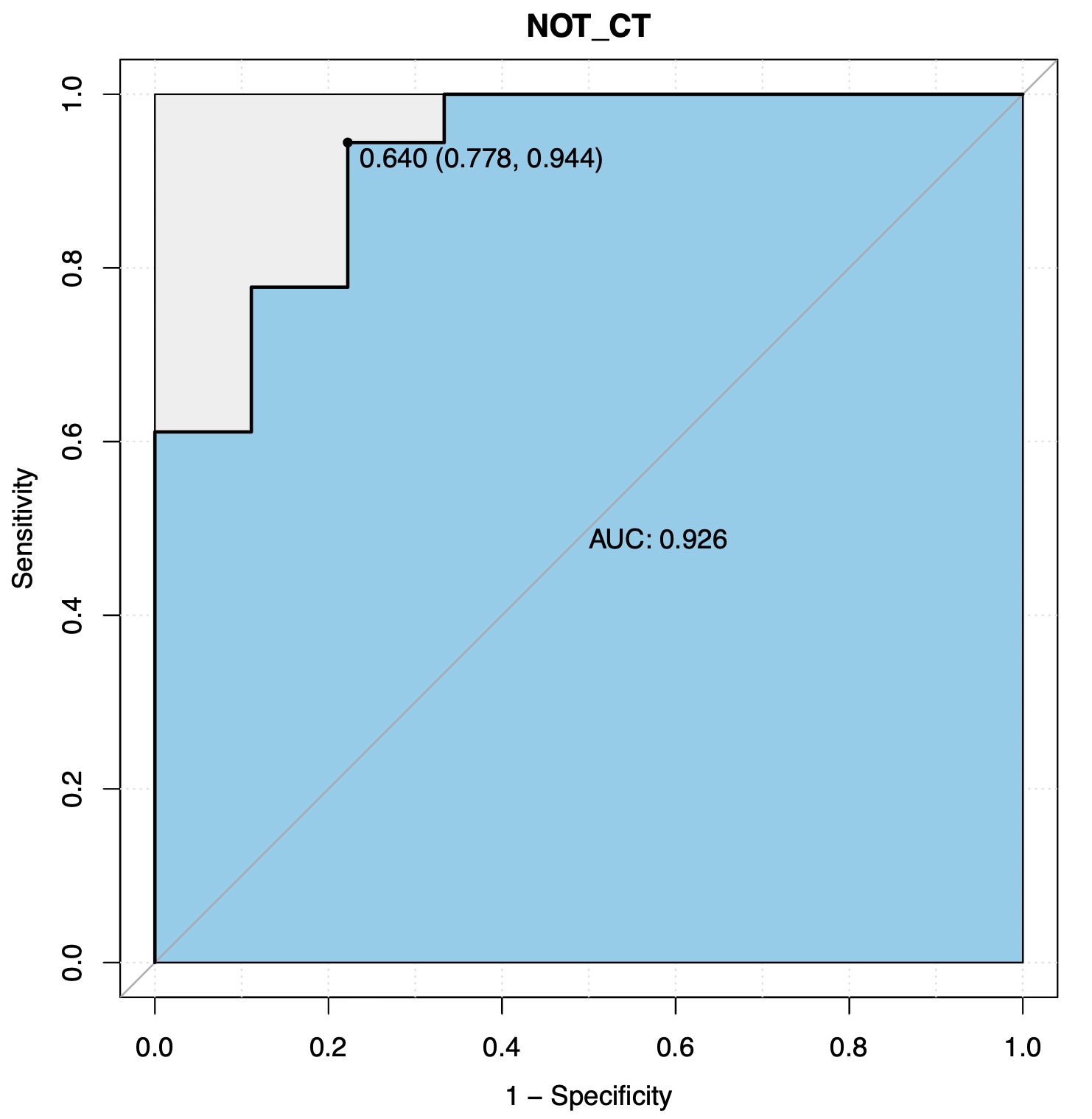
Tips 3: 随机森林模型在很多研究中被用作疾病筛查模型,因此在结果计算后需要采用ROC模型对随机森林的结果进行评估。
Tips 4: 如输入数据为二分类数据,例如高血压组和非高血压组,糖尿病组和非糖尿病组等,则无须下列转换步骤。
# 由于本示例数据为三组,分别为CT(正常饲料组),IO(铁剂过量饲料组)和ID(铁剂缺乏饲料组),无法直接使用与二分类的ROC模型评估
# 因此可以将数据转为二分类数据,例如CT组和非CT组
RF_data_binary <- RFCV_data_binary(RF_data,rf_estimate_group = 'CT',id_not = 'NOT_CT')
# 再次进行RFCV计算
RF_re <- RFCV(RF_data_binary)
# 将RFCV的结果直接带入函数
# rf_tax_select 为选取所需要进一步评估的物种,这里可以直接带入上并集结果
RFCV_roc(RF_re,rf_tax_select = RF_re$RFCV_result_plot$union_num,rf_estimate_group = 'NOT_CT')
# 也可以根据tax_plot函数的结果,手动挑选合适的物种纳入ROC评估
# 例如可以挑选出具有统计学差异的核心微生物物种
tax_re <- tax_plot(RF_data_binary,tax_select = RF_re2$RFCV_result_plot$union_num,method = 'ttest')
tax_re$pic$total
RFCV_roc(RF_re2,rf_tax_select = c('V83','V121','V143'),rf_estimate_group = 'NOT_CT')